Pulmonary hypertension
| Pulmonary hypertension | |
|---|---|
| Classification and external resources | |
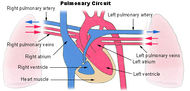 Pulmonary circuit |
|
| ICD-10 | I27.0, I27.2 |
| ICD-9 | 416 |
| DiseasesDB | 10998 |
| eMedicine | med/1962 |
| MeSH | D006976 |
In medicine, pulmonary hypertension (PH or PHT) is an increase in blood pressure in the pulmonary artery, pulmonary vein, or pulmonary capillaries, together known as the lung vasculature, leading to shortness of breath, dizziness, fainting, and other symptoms, all of which are exacerbated by exertion. Pulmonary hypertension can be a severe disease with a markedly decreased exercise tolerance and heart failure. It was first identified by Dr. Ernst von Romberg in 1891.[1] According to the most recent classification, it can be one of five different types: arterial, venous, hypoxic, thromboembolic or miscellaneous.[2]
Contents |
Signs and symptoms
Because symptoms may develop very gradually, patients may delay seeing a physician for years. Common symptoms are shortness of breath, fatigue, non-productive cough, angina pectoris, fainting or syncope, peripheral edema (swelling around the ankles and feet), and rarely hemoptysis (coughing up blood).
Pulmonary venous hypertension typically presents with shortness of breath while lying flat or sleeping (orthopnea or paroxysmal nocturnal dyspnea), while pulmonary arterial hypertension (PAH) typically does not.
A detailed family history is established to determine whether the disease might be familial. A history of exposure to drugs such as cocaine, methamphetamine, alcohol leading to cirrhosis, and tobacco leading to emphysema are considered significant. A physical examination is performed to look for typical signs of pulmonary hypertension, including a loud P2 (pulmonic valve closure sound), (para)sternal heave, jugular venous distension, pedal edema, ascites, hepatojugular reflux, clubbing etc. Evidence of tricuspid insufficiency is also sought and, if present, is consistent with the presence of pulmonary hypertension.
Diagnosis
Because pulmonary hypertension can be of five major types, a series of tests must be performed to distinguish pulmonary arterial hypertension from venous, hypoxic, thromboembolic, or miscellaneous varieties.
A physical examination is performed to look for typical signs of pulmonary hypertension. These include altered heart sounds, such as a widely split S2 or second heart sound, a loud P2 or pulmonic valve closure sound (part of the second heart sound), (para)sternal heave, possible S3 or third heart sound, and pulmonary regurgitation. Other signs include an elevated jugular venous pressure, peripheral edema (swelling of the ankles and feet), ascites (abdominal swelling due to the accumulation of fluid), hepatojugular reflux, and clubbing.
Further procedures are required to confirm the presence of pulmonary hypertension and exclude other possible diagnoses. These generally include pulmonary function tests; blood tests to exclude HIV, autoimmune diseases, and liver disease; electrocardiography (ECG); arterial blood gas measurements; X-rays of the chest (followed by high-resolution CT scanning if interstitial lung disease is suspected); and ventilation-perfusion or V/Q scanning to exclude chronic thromboembolic pulmonary hypertension. Biopsy of the lung is usually not indicated unless the pulmonary hypertension is thought to be due to an underlying interstitial lung disease. But lung biopsies are fraught with risks of bleeding due to the high intrapulmonary blood pressure. Clinical improvement is often measured by a "six-minute walk test", i.e. the distance a patient can walk in six minutes. Stability and improvement in this measurement correlate with better survival. Blood BNP level is also being used now to follow progress of patients with pulmonary hypertension.
Diagnosis of PAH requires the presence of pulmonary hypertension with two other conditions. Pulmonary artery occlusion pressure (PAOP or PCWP) must be less than 15 mm Hg (2000 Pa) and pulmonary vascular resistance (PVR) must be greater than 3 Wood units (240 dyn•s•cm-5 or 2.4 mN•s•cm-5).
Although pulmonary arterial pressure can be estimated on the basis of echocardiography, pressure measurements with a Swan-Ganz catheter provides the most definite assessment. PAOP and PVR cannot be measured directly with echocardiography. Therefore diagnosis of PAH requires right-sided cardiac catheterization. A Swan-Ganz catheter can also measure the cardiac output, which is far more important in measuring disease severity than the pulmonary arterial pressure.
Normal pulmonary arterial pressure in a person living at sea level has a mean value of 12–16 mm Hg (1600–2100 Pa). Pulmonary hypertension is present when mean pulmonary artery pressure exceeds 25 mm Hg (3300 Pa) at rest or 30 mm Hg (4000 Pa) with exercise.
Mean pulmonary artery pressure (mPAP) should not be confused with systolic pulmonary artery pressure (sPAP), which is often reported on echocardiogram reports. A systolic pressure of 40 mm Hg typically implies a mean pressure more than 25 mm Hg. Roughly, mPAP = 0.61•sPAP + 2.
Causes and classification
A 1973 meeting organized by the World Health Organization was the first to attempt classification of pulmonary hypertension. A distinction was made between primary and secondary PH, and primary PH was divided in the "arterial plexiform", "veno-occlusive" and "thromboembolic" forms.[3] A second conference in 1998 at Évian-les-Bains also addressed the causes of secondary PH (i.e. those due to other medical conditions),[4] and in 2003, the 3rd World Symposium on Pulmonary Arterial Hypertension was convened in Venice to modify the classification based on new understandings of disease mechanisms. The revised system developed by this group provides the current framework for understanding pulmonary hypertension.[2] The system includes several improvements over the former 1998 Evian Classification system. Risk factor descriptions were updated, and the classification of congenital systemic-to pulmonary shunts was revised. A new classification of genetic factors in PH was recommended, but not implemented because available data were judged to be inadequate.[2]
The Venice 2003 Revised Classification system can be summarized as follows:[2]
- WHO Group I - Pulmonary arterial hypertension (PAH)
- Idiopathic (IPAH)
- Familial (FPAH)
- Associated with other diseases (APAH): collagen vascular disease (e.g. scleroderma), congenital shunts between the systemic and pulmonary circulation, portal hypertension, HIV infection, drugs, toxins, or other diseases or disorders
- Associated with venous or capillary disease
- WHO Group II - Pulmonary hypertension associated with left heart disease
- Atrial or ventricular disease
- Valvular disease (e.g. mitral stenosis)
- WHO Group III - Pulmonary hypertension associated with lung diseases and/or hypoxemia
- Chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD)
- Sleep-disordered breathing, alveolar hypoventilation
- Chronic exposure to high altitude
- Developmental lung abnormalities
- WHO Group IV - Pulmonary hypertension due to chronic thrombotic and/or embolic disease
- Pulmonary embolism in the proximal or distal pulmonary arteries
- Embolization of other matter, such as tumor cells or parasites
- WHO Group V - Miscellaneous
The classification does not include sickle cell disease,[5] Human herpesvirus 8, also associated with Kaposi's sarcoma, has been demonstrated in patients with PAH, suggesting that this virus may play a role in its development.[6] Recent studies have been unable to find an association between human herpesvirus 8 and idiopathic pulmonary arterial hypertension.
Pathogenesis
Whatever the initial cause, pulmonary arterial hypertension (WHO Group I) involves the vasoconstriction or tightening of blood vessels connected to and within the lungs. This makes it harder for the heart to pump blood through the lungs, much as it is harder to make water flow through a narrow pipe as opposed to a wide one. Over time, the affected blood vessels become both stiffer and thicker, in a process known as fibrosis. This further increases the blood pressure within the lungs and impairs their blood flow. In addition, the increased workload of the heart causes hypertrophy of the right ventricle (a condition known as cor pulmonale), making the heart less able to pump blood through the lungs, ultimately causing right heart failure. As the blood flowing through the lungs decreases, the left side of the heart receives less blood. This blood may also carry less oxygen than normal. Therefore it becomes harder and harder for the left side of the heart to pump to supply sufficient oxygen to the rest of the body, especially during physical activity.
Pathogenesis in pulmonary venous hypertension (WHO Group II) is completely different. There is no obstruction to blood flow in the lungs. Instead, the left heart fails to pump blood efficiently, leading to pooling of blood in the lungs. This causes pulmonary edema and pleural effusions.
In hypoxic pulmonary hypertension (WHO Group III), the low levels of oxygen are thought to cause vasoconstriction or tightening of pulmonary arteries. This leads to a similar pathophysiology as pulmonary arterial hypertension.
In chronic thromboembolic pulmonary hypertension (WHO Group IV), the blood vessels are blocked or narrowed with blood clots. Again, this leads to a similar pathophysiology as pulmonary arterial hypertension.
Epidemiology
IPAH is a rare disease with an incidence of about 2-3 per million per year[7] and a prevalence of about 15 per million. Adult females are almost three times as likely to present with IPAH than adult males. The presentation of IPAH within children is more evenly split along gender lines.
Other forms of PAH are far more common. In scleroderma the incidence has been estimated to be 6 to 60% of all patients, in rheumatoid arthritis up to 21%, in systemic lupus erythematosus 4 to 14%, in portal hypertension between 2 to 5%, in HIV about 0.5%, and in sickle cell disease ranging from 20 to 40%.
Diet pills such as Fen-Phen produced an annual incidence of 25-50 per million per year.
Pulmonary venous hypertension is exceedingly common, since it occurs in most patients symptomatic with congestive heart failure.
Up to 4% of people who suffer a pulmonary embolism go on to develop chronic thromboembolic disease including pulmonary hypertension.
Only about 1.1% of patients with COPD develop pulmonary hypertension with no other disease to explain the high pressure. Sleep apnea is usually associated with only very mild pulmonary hypertension, typically below the level of detection. On the other hand Pickwickian syndrome (obesity-hypoventilation syndrome) is very commonly associated with right heart failure due to pulmonary hypertension.
Treatment
Treatment is determined by whether the PH is arterial, venous, hypoxic, thromboembolic, or miscellaneous. Since pulmonary venous hypertension is synonymous with congestive heart failure, the treatment is to optimize left ventricular function by the use of diuretics, beta blockers, ACE inhibitors, etc., or to repair/replace the mitral valve or aortic valve.
In PAH, lifestyle changes, digoxin, diuretics, oral anticoagulants, and oxygen therapy are considered conventional therapy, but have never been proven to be beneficial in a randomized, prospective manner.
High dose calcium channel blockers are useful in only 5% of IPAH patients who are vasoreactive by Swan-Ganz catheter. Unfortunately, calcium channel blockers have been largely misused, being prescribed to many patients with non-vasoreactive PAH, leading to excess morbidity and mortality. The criteria for vasoreactivity have changed. Only those patients whose mean pulmonary artery pressure falls by more than 10 mm Hg to less than 40 mm Hg with an unchanged or increased cardiac output when challenged with adenosine, epoprostenol, or nitric oxide are considered vasoreactive. Of these, only half of the patients are responsive to calcium channel blockers in the long term.
A number of agents has recently been introduced for primary and secondary PAH. The trials supporting the use of these agents have been relatively small, and the only measure consistently used to compare their effectivity is the "6 minute walking test". Many have no data on mortality benefit or time to progression.[8]
Vasoactive substances
Many pathways are involved in the abnormal proliferation and contraction of the smooth muscle cells of the pulmonary arteries in patients with pulmonary arterial hypertension. Three of these pathways are important since they have been targeted with drugs — endothelin receptor antagonists, phosphodiesterase type 5 inhibitors, and prostacyclin derivatives.
Because inexpensive generic drugs for this disease are not widely available, the World Health Organization does not include them in its model list of essential medicines.
Prostaglandins
Prostacyclin (prostaglandin I2) is commonly considered the most effective treatment for PAH. Epoprostenol (synthetic prostacyclin, marketed as Flolan) is given via continuous infusion that requires a semi-permanent central venous catheter. This delivery system can cause sepsis and thrombosis. Flolan is unstable, and therefore has to be kept on ice during administration. Since it has a half-life of 3 to 5 minutes, the infusion has to be continuous (24/7), and interruption can be fatal. Other prostanoids have therefore been developed. Treprostinil (Remodulin) can be given intravenously or subcutaneously, but the subcutaneous form can be very painful. An increased risk of sepsis with intravenous Remodulin has been reported by the CDC. Iloprost (Ilomedin) is also used in Europe intravenously and has a longer half life. Iloprost (marketed as Ventavis) is the only inhaled form of prostacyclin approved for use in the US and Europe. This form of administration has the advantage of selective deposition in the lungs with less systemic side effects. Oral and inhaled forms of Remodulin are under development. Beraprost is an oral prostanoid available in Japan and South Korea.
Endothelin receptor antagonists
The dual (ETA and ETB) endothelin receptor antagonist bosentan (marketed as Tracleer) was approved in 2001. Sitaxentan, a selective endothelin receptor antagonist that blocks only the action of ETA, has been approved for use in Canada, Australia, and the European Union, to be marketed under the name Thelin.[9] Sitaxentan has not been approved for marketing by the U.S. Food and Drug Administration (FDA). A new trial to address the FDA's concerns will begin in 2008. A similar drug, ambrisentan is marketed as Letairis in U.S. by Gilead Sciences.[10] In addition, another dual/nonselective endothelin antagonist, Actelion-1, from the makers of Tracleer, will enter clinical trials in 2008.
Phosphodiesterase type 5 inhibitors
Sildenafil, a selective inhibitor of cGMP specific phosphodiesterase type 5 (PDE5), was approved for the treatment of PAH in 2005. It is marketed for PAH as Revatio.
Activators of soluble guanylate cyclase
Soluble guanylate cyclase (sGC) is the intracellular receptor for NO. As of April 2009[update], the sGC activators cinaciguat and riociguat are undergoing clinical trials for the treatment of PAH.
Surgical
Atrial septostomy is a surgical procedure that creates a communication between the right and left atria. It relieves pressure on the right side of the heart, but at the cost of lower oxygen levels in blood (hypoxia).
Lung transplantation cures pulmonary arterial hypertension, but leaves the patient with the complications of transplantation, and a post-surgical median survival of just over five years.[11]
Pulmonary thromboendarterectomy (PTE) is a surgical procedure that is used for chronic thromboembolic pulmonary hypertension. It is the surgical removal of an organized thrombus (clot) along with the lining of the pulmonary artery; it is a very difficult, major procedure that is currently performed in a few select centers. Case series show remarkable success in most patients.
Treatment regimens for hypoxic and miscellaneous varieties of pulmonary hypertension have not been established. However, studies of several agents are currently enrolling patients. Many physicians will treat these diseases with the same medications as for PAH, until better options become available. Such treatment is called off-label use.
Monitoring
Patients are normally monitored through commonly available tests such as:
- pulse oximetry,
- arterial blood gas tests,
- chest X-rays,
- serial ECG tests,
- serial echocardiography, and
- spirometry or more advanced lung function studies.
Prognosis
The NIH IPAH registry from the 1980s showed an untreated median survival of 2–3 years from time of diagnosis, with the cause of death usually being right ventricular failure (cor pulmonale). Although this figure is widely quoted, it is probably irrelevant today. Outcomes have changed dramatically over the last two decades. This may be because of newer drug therapy, better overall care, and earlier diagnosis (lead time bias). A recent outcome study of those patients who had started treatment with bosentan (Tracleer) showed that 89% patients were alive at 2 years.[12] With multiple agents now available, combination therapy is increasingly used. Impact of these agents on survival is not known, since many of them have been developed only recently. It would not be unreasonable to expect median survival to extend past 10 years in the near future.[13]
Levels of mortality are very high in pregnant women with severe pulmonary hypertension.[14] Pregnancy is sometimes described as contraindicated in these women.[15][16][17]
See also
- Pulmonary Hypertension Association
- Risk factors in pregnancy
References
- ↑ von Romberg, Ernst (1891-1892). "Über Sklerose der Lungenarterie" (in German). Dtsch Arch Klin Med 48: 197–206.
- ↑ 2.0 2.1 2.2 2.3 Simonneau G, Galiè N, Rubin LJ, et al. (June 2004). "Clinical classification of pulmonary hypertension". J. Am. Coll. Cardiol. 43 (12 Suppl S): 5S–12S. doi:10.1016/j.jacc.2004.02.037. PMID 15194173. http://content.onlinejacc.org/cgi/content/full/43/12_Suppl_S/5S.
- ↑ Hatano S, Strasser R (1975). Primary pulmonary hypertension. Geneva: World Heath Organization.
- ↑ Rich S, Rubin LJ, Abenhail L et al. (1998). Executive summary from the World Symposium on Primary Pulmonary Hypertension (Evian, France, September 6–10, 1998). Geneva: The World Health Organization. http://web.archive.org/web/20020408173726/http://www.who.int/ncd/cvd/pph.html.
- ↑ Gladwin MT, Sachdev V, Jison ML, et al. (2004). "Pulmonary hypertension as a risk factor for death in patients with sickle cell disease". N. Engl. J. Med. 350 (9): 886–95. doi:10.1056/NEJMoa035477. PMID 14985486. http://content.nejm.org/cgi/content/full/350/9/886.
- ↑ Cool CD, Rai PR, Yeager ME, et al. (2003). "Expression of human herpesvirus 8 in primary pulmonary hypertension". N. Engl. J. Med. 349 (12): 1113–22. doi:10.1056/NEJMoa035115. PMID 13679525. http://content.nejm.org/cgi/content/full/349/12/1113.
- ↑ Rudarakanchana, N; Trembath RC, Morrell NW (November 2001). "New insights into the pathogenesis and treatment of primary pulmonary hypertension". Thorax 56 (11): 888–890. doi:10.1136/thorax.56.11.888. PMID 11641516.
- ↑ Torres F (2007). "Systematic review of randomised, double-blind clinical trials of oral agents conducted in patients with pulmonary arterial hypertension". Int. J. Clin. Pract. 61 (10): 1756–65. doi:10.1111/j.1742-1241.2007.01545.x. PMID 17877662. http://www.blackwell-synergy.com/doi/full/10.1111/j.1742-1241.2007.01545.x.
- ↑ "UPDATE 1-Encysive gets Canadian approval for hypertension drug". Reuters. 2008-05-30. http://www.reuters.com/article/governmentFilingsNews/idUSBNG28335020070530. Retrieved 2007-07-08.
- ↑ Gilead Sciences (2007-06-15). "U.S. Food and Drug Administration Approves Gilead's Letairis Treatment of Pulmonary Arterial Hypertension". Press release. http://www.gilead.com/wt/sec/pr_1016053. Retrieved 2007-06-16.
- ↑ "2006 OPTN/SRTR Annual Report". US Scientific Registry of Transplant Recipients. 2006-05-01. http://www.ustransplant.org/annual_reports/current/113_surv-new_dh.htm. Retrieved 2007-03-28.
- ↑ McLaughlin VV, Sitbon O, Badesch DB, et al. (2005). "Survival with first-line bosentan in patients with primary pulmonary hypertension". Eur. Respir. J. 25 (2): 244–9. doi:10.1183/09031936.05.00054804. PMID 15684287. http://erj.ersjournals.com/cgi/content/full/25/2/244.
- ↑ Nauser TD, Stites SW (2001). "Diagnosis and treatment of pulmonary hypertension". Am Fam Physician 63 (9): 1789–98. PMID 11352291. http://www.aafp.org/afp/20010501/1789.html.
- ↑ Kelley's essentials of internal medicine. Hagerstwon, MD: Lippincott Williams & Wilkins. 2001. pp. 84. ISBN 0-7817-1937-2.
- ↑ Edward Benz; David Weatherall; David Warrell; Cox, Timothy J.; Firth, John B. (2005). Oxford textbook of medicine. Oxford [Oxfordshire]: Oxford University Press. pp. 1101. ISBN 0-19-856978-5.
- ↑ Kaufman, Matthew H.; Latha Stead; Feig, Robert (2007). First aid for the obstetrics & gynecology clerkship. New York: McGraw-Hill, Medical Pub. Division. pp. 100. ISBN 0-07-144874-8.
- ↑ Ghosh, Amit K. (2008). Mayo Clinic Internal Medicine Review: Eighth Edition (Mayo Clinic Internal Medicine Review). Informa Healthcare. pp. 55. ISBN 1-4200-8478-X.
Sources
- Rubin LJ, Badesch DB (2005). "Evaluation and management of the patient with pulmonary arterial hypertension". Ann. Intern. Med. 143 (4): 282–92. PMID 16103472. http://www.annals.org/cgi/reprint/143/4/282.
External links
- The Merck Manual of Diagnosis and Therapy: Pulmonary Hypertension
- Pulmonary Hypertension Association
- PH Central - the internet resource for Pulmonary Arterial Hypertension
- Facts About Primary Pulmonary Hypertension from the National Heart, Lung, and Blood Institute (NHLBI)
- Webcast: The Changing World of Pulmonary Arterial Hypertension Therapies - American College of CHEST Physicians
- pph at NIH/UW GeneTests - "BMPR2-Related Primary Pulmonary Hypertension"
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||